Introduction
Mental retardation, X-linked 90 (MRX90), (OMIM300850) caused by mutation on DLG3, up to date only 14 DLG3 mutations have been reported. the male patients had clinical phenotype range from moderate to severe Intellectual Disability (ID) and dysmorphic features include Strabismus, up-slanting palpebral fissures, high-arched palate, and narrow thorax the majority female heterozygous mutation carriers was asymptomatic and had random X-inactivation patterns. In recent report of clinical phenotypical of DLG3 mutation caused XLID and demonstrates that heterozygous female mutation carriers can be as severely affected as males [1].
X-Linked Intellectual Disability (XLID) is a highly heterogeneous disorder, accounts for 5%-10% of intellectual disability in males. About 150 syndromes were described [2] and more than 102 genes identified up to date.
The Disc-Large homolog 3 (DLG3) gene, encoding the synapse-associated protein 102 (SAP102) which belongs to the Membrane-Associated Guanylate Kinases (MAGUK) protein [3]. Neuronal SAP102 is expressed during early brain development and is localized to the postsynaptic density of excitatory synapses [4].
Mutations in DLG3 are a rare cause of non-syndromic XLID (MRX90, OMIM #300850); only 21 Patients have been reported to date (HGMD). The male affected with variable degree of Cognitive impairment and with other clinical finding including seizures, abnormal behavior, and facial dysmorphisms [5,6].
Most female carriers of heterozygous DLG3 mutations were asymptomatic or mildly affected, due to skewed X-inactivation, rarely severely affected. The Laura Gieldon and his colleagues [7], have been reported five females with variable cognitive impairment and in one of these patients, who was suffering from moderate intellectual disabilities and random X-inactivation. Here, we provide a description of one Saudi female with Mental retardation, X-linked 90 (MRX90), associated with a novel mutation in DLG3 gene. And It showed the severity of clinical phenotypic in female carrier correlates with the X-inactivation status.
Clinical Report
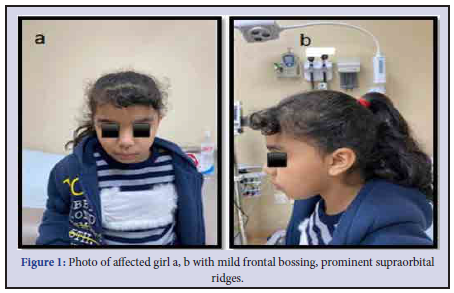
The patient, a 10-year-old girl, product of consanguineous Saudi parents. and have three other healthy children She was born after an uneventful pregnancy after 39 weeks’ gestation with the following birth parameters: weight 3,715 g (±0 SD), length 53 cm (+0.5 SD), and head circumference 34 cm (–1 SD). No antenatal or neonatal complained. She was referred to genetic clinic with history of presented with moderate ID, an attention deficit, and hyperactive behavior, poor concentration and learning disabilities attend special school for difficult learning. No of history seizure or abnormal movements. She was started to crawl at age of 1 years and 3 months and she apparently started to walk without support at the age of 2 years and 8 months. She had delayed speech development. At the age of 5 years, she spoke 4-6 words, and at 9 years of age, she only had spoken 3-word phrases. At the time of evaluation at 8 years of age, her height was 126 cm (10 SD), weight was 25 kg (25 SD), and head circumference was 51 cm (25 SD). She had mild frontal bossing, prominent supraorbital ridges (Figure 1) with normal hands, feet, toes, no chest deformities, and the rest of examination were normal. Magnetic Resonance Imaging (MRI) of the brain at 8 years of age was unremarkable study. Conventional chromosome analysis was normal female karyotype.
Material and Methods
Double stranded DNA capture baits against approximately 36.5 Mb of the human coding exome (targeting >98% of the coding RefSeq from the human genome build GRCh37/hg19) are used to enrich target regions from fragmented genomic DNA with the Twist Human Core Exome Plus kit. The generated library is sequenced on an Illumina platform to obtain at least 20x coverage depth for >98% of the targeted bases. An in-house bioinformatics pipeline, including read alignment to GRCh37/hg19 genome assembly, variant calling (single nucleotide and small deletion/insertion variants), annotation and comprehensive variant filtering is applied. All variants with Minor Allele Frequency (MAF) of less than 1% in gnomAD database, and disease-causing variants reported in HGMD®, in ClinVar or in CentoMD® are considered. The investigation for relevant variants is focused on coding exons and flanking +/-20 intronic nucleotides of genes with a clear gene-phenotype evidence (based on OMIM® information). All potential modes of inheritance patterns are considered. In addition, provided family history and clinical information are used to evaluate identified variants with respect to their pathogenicity and causality.
Results
Whole exome sequencing showed a novel heterozygous frameshift mutation c.1695del p. (Arg566Glyfs*10) was detected. Whole exome sequence Solo confirmed this mutation in both patients. Both parents testing was normal for this mutation. Which confirmed this mutation is de novo.
The mutation c.1695del p.(Arg566Glyfs*10) is novel, Similarly, this heterozygous variant was not found in 200 controls, 1000G, and ExAC database. According to the ACMG Standards and Guidelines, this variant was classified as “likely pathogenic with clinical phenotype and confirmed mutation that supports the hypothesis that this mutation is responsible for a skewed inactivation of X, and that only the normal X can be inactivated. Therefore, the female patient was diagnosed as a manifesting DLG3 carrier.
Discussion
We report a family, a girl, with a novel nonsense gene variant of DLG3, c.1695del p. (Arg566Glyfs*10). She showed ID, hyperactive behavior, dysmorphic features (Figure 1) and normal had brain MRI.
The Anna Sandestig [6] described mother of index case boy with DLG3 mutation had learning disabilities and a borderline cognitive development and no other clinical features. Her disease-causing allele had arisen de novo.
In X-linked intellectual disability, which is heterogeneous, the affected gene is maternally inherited and the males are at higher risk to inherit the defect from the carrier mother and usually have severe phenotype. This is explaining the severity of ID in males compare to females. And females can express the defect in a recessive manner due to skewed X-inactivation [9].
X-linked mental retardation-90 (MRX90) is caused by mutation in the DLG3. It is a heterogeneous disorder with phenotype spectrum sever to moderate ID. It was reported in affected patients most of them are men with nonsyndromic ID, ranging from mild to severe, abnormal behavior and seizure which were previously published patients with DLG3mutations [1,3,8], and some of the patients had distinctive dysmorphic features have been reported as upslanting palpebral fissures, triangular face, high-arched palate, narrow thorax, and Broad great toes have been reported [10].
Most of the case were reported affected males but the female affected has been reported [8], see table 1, with severely clinical phenotype like male presentation and this was due to skewed X-inactivation - the normal DLG3 allele was inactive. They showed that the severity of phenotypic expression in female carriers correlates with the X-inactivation status.
Conclusion
We identified de novo mutations in DLG3 in one Saudi girl ID, abnormal behavior and without a relevant family history, and WES is an effective approach to reached diagnosis in ID, particularly in female patient without a relevant family history, to detect unexpected DNA variations.
Acknowledgements
We are grateful to the patient and their family and CENTOGENE GmbH (Rostock, Germany). For their dedicated assistance and support.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.